Updates
Unveiling TREAT-1: Advanced Binding Predictions for DAT, SERT, AND NET
POSTED ON
May 7, 2025

We recently launched TREAT (Targeted Reward Evaluation and Therapeutics), an initiative focused on mapping the neural pathways that govern reward, learning, and bodily rhythms.
Today, we’re excited to unveil TREAT-1, our first fine-tuned model designed to target key monoamine transporters—dopamine (DAT), serotonin (SERT), and norepinephrine (NET). These transporters play a pivotal role in mood, cognition, and attention.
TREAT-1 builds on the PSICHIC architecture, fine-tuned to deliver superior predictive power and unlock new insights into transporter-ligand interactions. This step forward isn't just about precision, it's about accelerating the discovery of therapeutics that target neurological and psychiatric conditions at their core.
But TREAT-1 is just the start. Powered by NOVA (#Bittensor #SN68), our decentralized AI platform, we are on track to push the boundaries of drug discovery and support the development of neuromodulatory therapeutics with greater precision.
Summary
Protein–ligand affinity prediction is crucial for accelerating drug discovery, but most leading models depend on scarce and expensive 3D structural data.
PSICHIC: A state-of-the-art, sequence-only graph-attention network that incorporates physicochemical constraints, offering a structure-agnostic alternative is highly versatile.
But, PSICHIC’s performance on specialized transporter targets like dopamine (DAT), serotonin (SERT), and norepinephrine (NET) required further refinement.
TREAT-1: The first fine-tuned model from Metanova Labs' TREAT (Targeted Reward Evaluation and Therapeutics) initiative, designed to significantly enhance predictive accuracy for these key targets.
Model and trained weights for TREAT-1 are available on Hugging Face.
TREAT-1 demonstrates +79% enrichment factor compared to the baseline PSICHIC model, paving the way for more accurate and efficient drug discovery targeting critical neurotransmitter systems.
Introduction
Understanding and accurately modeling how small-molecule ligands interact with their protein targets is central to modern drug discovery and chemical biology. These interactions dictate pharmacological efficacy and selectivity, drive off-target liabilities, and steer the prioritization of compounds for costly experimental validation. Reliable in-silico models can therefore compress discovery timelines and reduce laboratory expense by focusing experimental effort on candidates with the greatest chance of success.
Over the past two decades, three broad families of computational techniques have emerged for protein–ligand modeling:
Structure-based approaches which rely on experimentally determined or in-silico protein structure, allowing the model to reason explicitly about the 3d geometry of the binding pocket. While this extra spatial context often improves accuracy, it comes at a price: high-resolution crystal or cryo-EM structures are expensive and scarce; in-silico models such as AlphaFold can introduce coordinate errors or capture an irrelevant conformational state; and performance degrades as structural resolution worsens.
Complex-based methods that take the entire co-crystallised protein–ligand complex as input, letting the model exploit precise 3d contact geometry and often yielding the highest predictive accuracy, but they depend on scarce, costly high-resolution complexes and break down when such data are unavailable.
Sequence-based methods, by contrast, which dispense entirely with 3d coordinates and operate on one- or two-dimensional inputs: protein primary sequences, contact-map predictions, and ligand SMILES or molecular graphs. Because they require only readily available sequence information, they avoid the cost, scarcity, and resolution-related pitfalls of structure-based pipelines, and they remain agnostic to the particular protein conformation captured experimentally or computationally. However, the lack of physicochemical constraints can cause sequence-based models to suffer from unrestrained degrees of freedom, which may cause the model to learn patterns that are not relevant to the interaction with small molecules, hindering its generalizability.
PSICHIC (PhySIcoCHemICal graph neural network) is a state-of-the-art algorithm that offers a fourth, sequence-only path that closes, or even reverses, the historical performance gap. By embedding explicit physicochemical constraints inside a cross-attention GNN architecture, PSICHIC learns interpretable “interaction fingerprints” directly from sequence data. In benchmark studies on PDBBind-2016/2020 it matched or surpassed leading structure- and complex-based models in affinity prediction, while remaining agnostic to structural resolution and generating residue- and atom-level importance maps that align with experimentally known binding sites.
These properties make PSICHIC an attractive starting point for domain-specific fine-tuning: its strong out-of-the-box accuracy for a variety of targets, reliance solely on readily available sequences and SMILES, and intrinsic interpretability provide a robust foundation for adapting the model to particular protein families, assay formats, or chemical spaces. The remainder of this report details the data curation, training protocol, and empirical results obtained when fine-tuning PSICHIC for our target application, TREAT.
TREAT (Targeted Reward Evaluation And Therapeutics) is an initiative aimed at identifying drug candidates to modulate and treat conditions that affect neural pathways regulating learning, mood and bodily rhythms, such as sleep and appetite. Some of the neurotransmitter systems that are relevant for these and many other processes are dopamine, serotonin, and norepinephrine, i.e., the monoamines. TREAT-1, the first domain-specific model to be deployed at NOVA (#Bittensor subnet 68) has been fine-tuned with data on the monoamine transporters, carefully curated from the literature, patents and other scientific publications.
After the monoamines deliver their signals in the synaptic cleft, three dedicated transport proteins— the dopamine transporter (DAT), serotonin transporter (SERT), and norepinephrine transporter (NET)—quickly vacuum the molecules back into the sending nerve cell. This resets the signal for the next nerve impulse and helps keep neurotransmitter levels in balance. Because these proteins gate pathways that underlie reward, mood and attention, they are prime drug-discovery targets: psychostimulants such as methylphenidate block DAT and NET, selective-serotonin-reuptake inhibitors (SSRIs) trap SERT, and the ADHD medicine atomoxetine selectively inhibits NET. Altered transporter expression or polymorphisms have been linked to Parkinson’s disease, major depression and ADHD, underscoring the need for subtype-selective modulators and a deeper structural understanding of transporter–ligand recognition.
Methods and Results
Fine-tuning strategy
We began with the multitask version of PSICHIC, which had been pre-trained on a large-scale protein–ligand interaction corpus that annotates both interaction type (binder, agonist, etc.) and strength.
Two fine-tuning approaches were evaluated:
Domain-specific fine-tuning (DS-FT). • From the original corpus we isolated all records for the proteins of interest (≈ 11 k protein–ligand pairs). • Training was resumed with a reduced learning rate. • Unlike the original study, which updated only the output layer, we unfroze and optimized the final three modules. Preliminary experiments showed that this deeper adaptation yielded systematically better performance for our target panel.
Domain-specific fine-tuning with data augmentation (DS-FT + A). • We augmented the domain-specific set with 3.7k new affinity measurements for human and rat DAT, NET, and SERT, manually curated from recent literature. • The augmented data were partitioned into five Bemis–Murcko scaffold folds, ensuring every chemical scaffold appeared in only one fold and eliminating scaffold leakage. • For each fold, compounds in the held-out fold served as an external validation set while the remaining four folds were used for fine-tuning, encouraging generalization to novel scaffolds.
Evaluation
The baseline model and both fine-tuned variants were assessed on the five external validation folds. The following performance metrics were used for their evaluatIon:
Mean Absolute Percentage Error (MAPE) is a metric that calculates the average percentage difference between predicted values and actual values. It’s useful for evaluating the accuracy of regression models by providing a straightforward percentage of error, making it easy to interpret. MAPE is commonly used in the fine-tuning process to track improvements in model performance. Lower MAPE values indicate that the model’s predictions are becoming more accurate. It helps assess whether adjustments made during fine-tuning are improving the model’s ability to generalize to new data.
Coefficient of determination (R²) is a statistical measure that indicates how well the predicted values from a model match the actual values. It represents the proportion of the variance in the dependent variable that is predictable from the independent variables. R² is used in fine-tuning to assess how well the model is fitting the data, with higher values indicating better fit and greater predictive accuracy.
F-1 score is a metric that combines precision and recall into a single number, providing a balance between the two. It’s particularly useful for evaluating models on imbalanced datasets, where one class is more frequent than the other. The F-1 score is used during fine-tuning to ensure that the model not only classifies correctly but also maintains a good balance between false positives and false negatives.
Enrichment factor measures how much better a model performs in identifying true positive instances compared to random selection. It calculates the ratio of true positives in the top-ranked predictions versus the total number of positives in the dataset. The enrichment factor is key for evaluating fine-tuning in drug discovery or similar fields, as it highlights the model's ability to prioritize high-quality results from large datasets.
Mean Absolute Percentage Error (MAPE) and coefficient of determination (R²) between experimental and predicted affinities are summarized in Figures 1 and 2.

Fig. 1: MAPE between real and predicted values in the baseline and fine-tuned versions of the model.
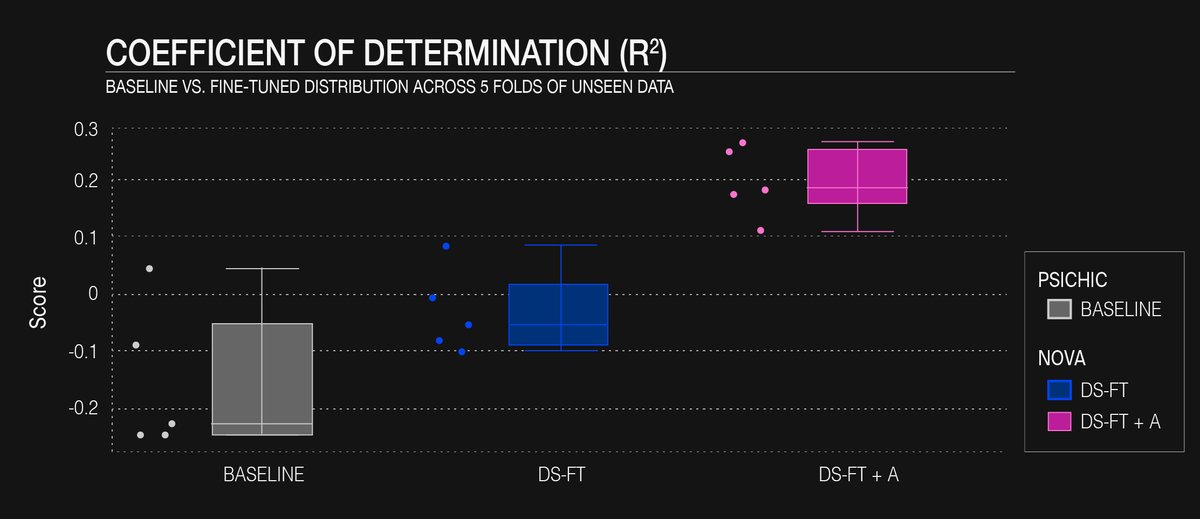
Fig. 2: R² between real and predicted values in the baseline and fine-tuned versions of the model.
For the DAT/NET/SERT panel the unhoned, multitask PSICHIC model produced a MAPE of ≈ 0.50 and mostly negative or near-zero R² on every fold (Fig. 1 and 2), confirming that it failed to capture the variance of these specific targets.
Updating the final three transformer blocks with the domain-specific slice (“DS-FT”) trimmed the MAPE by ≈ 10 % and nudged the mean R² barely into positive territory.
When the curated set of 3.7 k new affinity measurements was added (“DS-FT + A”) the improvement became substantive: the MAPE fell by a further ≈ 20 % and the mean R² climbed to ~ 0.2 across the five scaffold-held-out folds (Fig. 2).
These gains highlight a recurring theme in chemoinformatics: a comparatively small, high-quality supplement can outweigh lengthy re-training on broader but less relevant data.
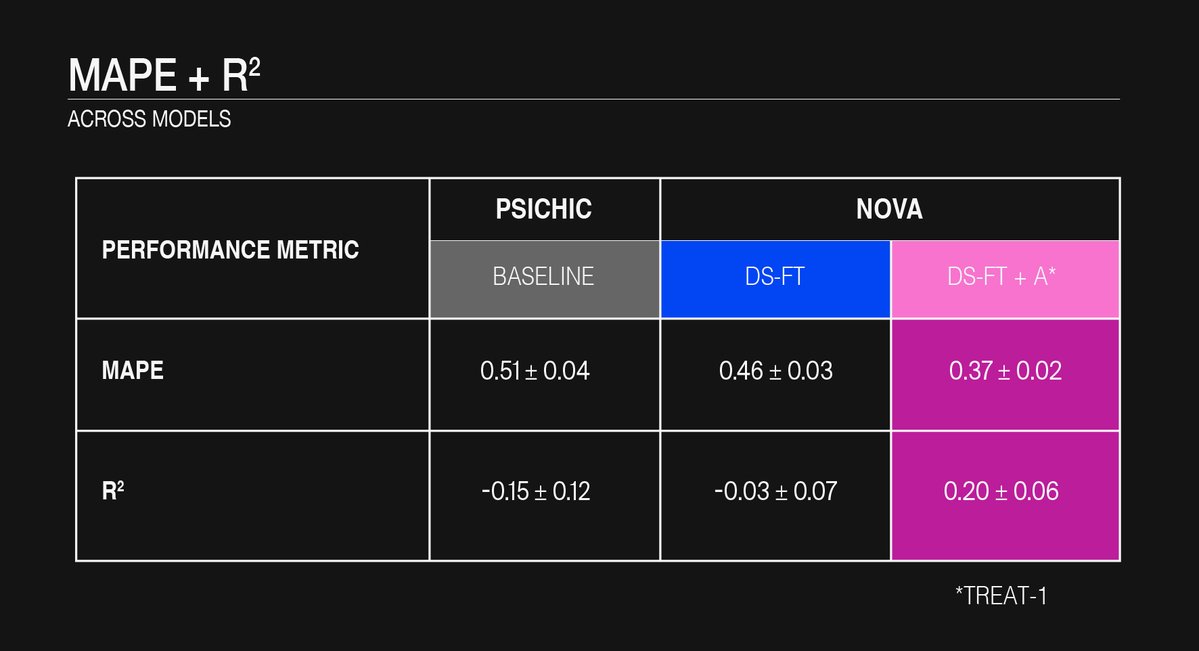
Table 1: MAPE and R² across models.
Using the regression model as a virtual-screening classifier
Because PSICHIC outputs continuous pKi/pIC₅₀-like scores, it doubles as a binary classifier without architectural changes:
Choose a biochemical cut-off. For example, pKi ≥ 6.5 defines an “active” binder.
Leave training untouched. Continue to fit the model on the original regression targets.
Apply an inference-time threshold. If the predicted score ŷ ≥ 6.5 label the compound active; otherwise inactive.
Compute classification statistics (precision, recall, enrichment factor, etc.) while still retaining the ranking resolution of the regression output.
Practical enrichment
Figure 3 illustrates this post-hoc classification for the human SERT subset (UniProt P31645):
The pink line marks the experimental activity threshold (pKi = 6.5).
The blue line applies the same threshold to the model predictions.
Excluding compounds below the blue line eliminates 60% of the candidates yet preserves most true actives, lifting the active fraction from 39.5 % in the original set to 70% in the retained subset, which means an enrichment factor of 1.79 × (≈ + 79%).
Such selective triage is invaluable when downstream assay throughput is limited, allowing wet-lab screens to focus on a much higher proportion of likely binders while still benefiting from the nuanced ranking that the regression model provides.
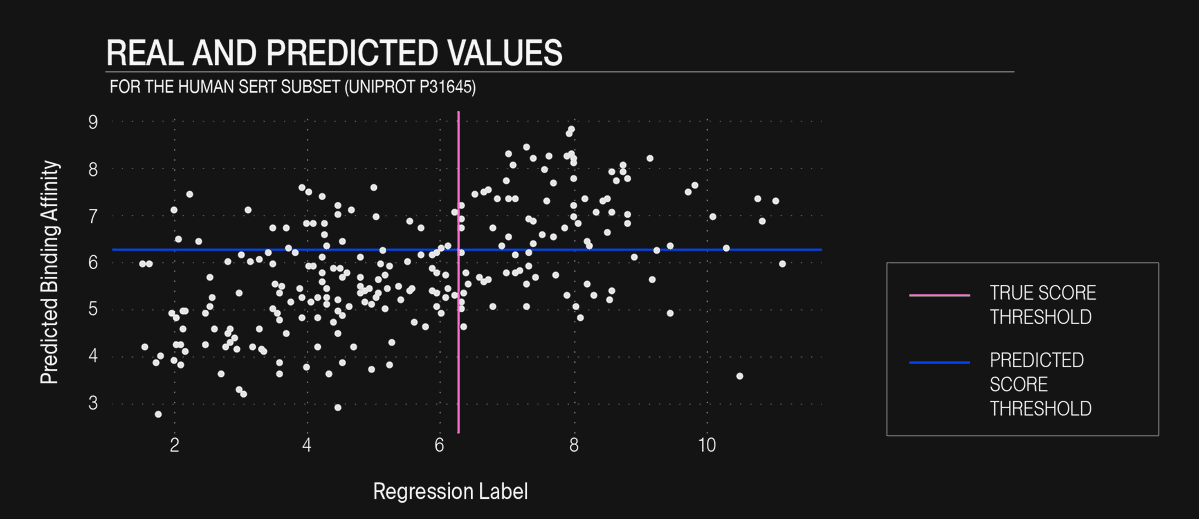
Fig. 3: Real and predicted values for one held-out set of the human SERT. Pink line represents the true score threshold to consider a sample as a binder and the blue line represents the predicted score threshold to label binders and non-binders.
Figure 4 compares F1-score for positive and negative classes across models for the five held-out folds and figure 4 compares enrichment factor. Both the biochemical cut-off and inference-time threshold were set to 6.5, which was the value that resulted in the highest enrichment factor for all models.
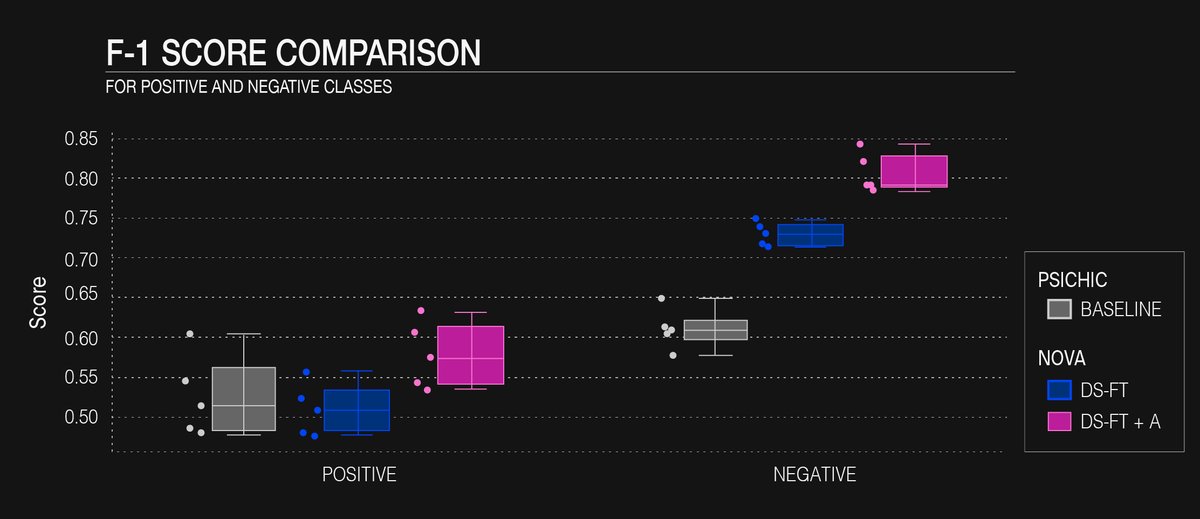
Fig. 4: F1-Score for positive and negative classes for each held-out fold across models.

Fig. 5: Enrichment factor for each held-out fold across all models.
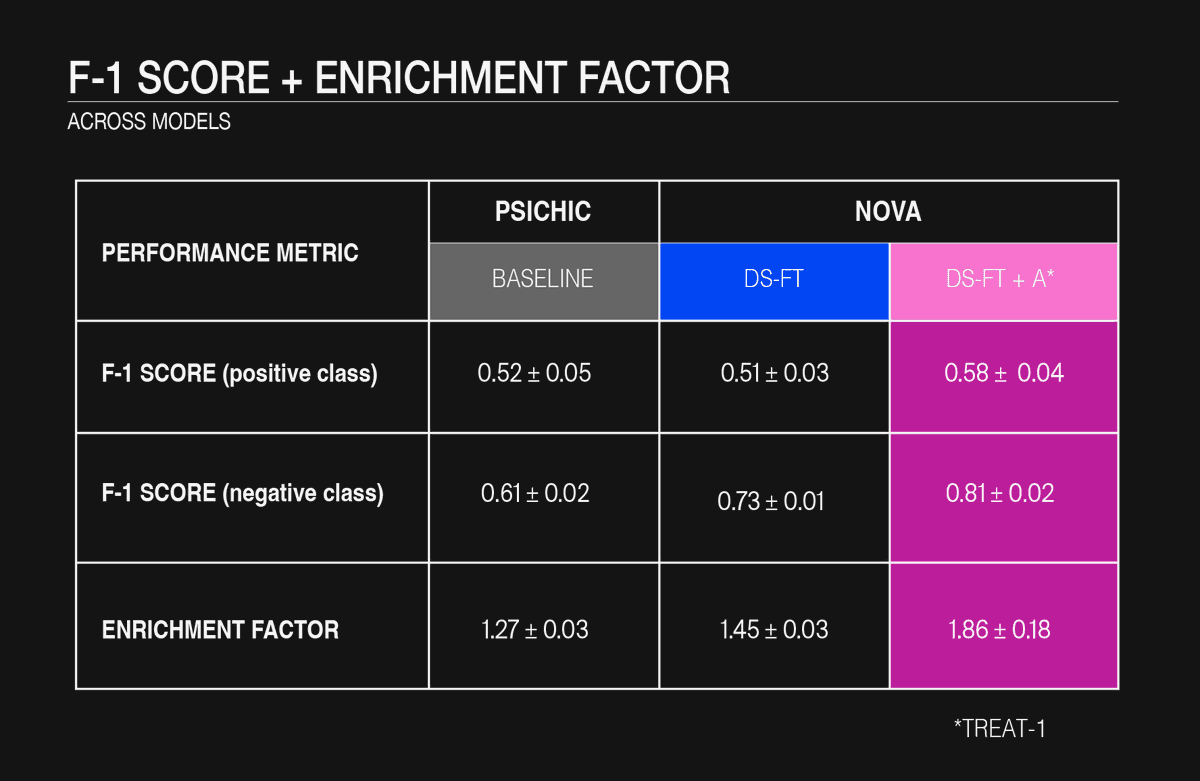
Table 2: F-1 score and Enrichment Factor across models
Across all metrics, the baseline model provides a useful lower bound but leaves substantial room for improvement: its median F1-score is roughly 0.52 for the positive (binder) class and 0.61 for the negative class, and its enrichment factor sits just above 1.2, so it identifies only a modest excess of true binders over random chance. Domain-specific fine-tuning (DS-FT) substantially sharpens the model’s ability to reject non-binders: negative-class F1 rises to about 0.73 and the enrichment factor to ~1.45. Yet, this gain comes at the cost of a slight dip in positive-class F1, which falls to around 0.50, indicating some loss of sensitivity to genuine actives. Augmenting the fine-tuning data with 3.7 k carefully curated, scaffold-split affinity records (DS-FT + A) produces the strongest overall performance: median F1 climbs to roughly 0.58 for positives and 0.81 for negatives, while the enrichment factor approaches 1.75, representing a 40% boost over DS-FT and nearly 70% over the baseline. Although the augmented model exhibits a wider fold-to-fold spread, an expected consequence of isolating chemical scaffolds, it achieves the best balance between filtering out inactives and recovering new actives, making it the preferred choice when maximizing hit discovery outweighs concerns about modest variability.
Conclusion
This study demonstrates that PSICHIC, a sequence-only graph-attention architecture, can be effectively repurposed for highly specialised transporter targets through fine-tuning and targeted data augmentation. Starting from a multitask model that performed little better than chance on DAT, SERT and NET, we showed that (i) unfreezing a few terminal blocks and continuing training on ~11 000 in-domain examples (DS-FT) already cuts error by one-tenth and meaningfully improves scaffold-held-out generalisation, and that (ii) supplementing this corpus with just 3700 high-quality, scaffold-partitioned affinities (DS-FT + A) delivers a further 20% reduction in MAPE, lifts mean R² to ~0.2, and—critically for virtual screening—raises the enrichment factor to ~1.75 while maintaining strong, balanced F1-scores for both actives and inactives. Because the regression output can be thresholded at inference time, the same model seamlessly doubles as a classifier, enabling rapid triage that nearly doubles the proportion of true binders in a filtered subset and thereby compresses downstream assay effort.
These gains underscore two key principles for chemoinformatics pipelines: first, depth-wise fine-tuning of a well-regularised backbone can unlock target-family signal that is otherwise invisible to shallow transfer; and second, a modest quantity of carefully curated, scaffold-diverse data often outweighs far larger but noisier corpora. Within Metanova Labs’ TREAT initiative, the DS-FT + A model is now positioned to prioritise compounds for synthesis and cellular profiling, with particular promise for uncovering transporter-selective leads that escape structure-based docking due to limited or low-resolution transporter conformations.
Next Steps
The launch of the TREAT-1 model marks just the beginning of our journey. Moving forward, we’ll focus on refining the model for enhanced selectivity, incorporating active learning loops, and deepening mechanistic interpretability to unlock even more profound biological insights. These advancements will drive the curation of novel chemical libraries and the selection of top compounds for synthesis and validation.
TREAT-1 will soon be deployed within Metanova Labs' NOVA (SN68). This is just the first step toward revolutionizing how we discover and develop life-changing therapeutics.
Join us today!
Mine, validate & stake $TAO to power the network. Everyone can play a role in advancing biomedical technology.